Malignant rhabdoid tumor (MRT) is one of the most aggressive and lethal childhood cancers.
Although rare in the United States, approximately 20 to 25 new cases are diagnosed each year, but the disease lacks standard and effective treatment because the patient has lost an anti-cancer protein called SNF5. A child with MRT is unlikely to survive a year after diagnosis.
Researchers at Vanderbilt University have found that a proto-oncogene-encoded protein, MYC, is often inhibited by SNF5. The absence of SNF5 is equivalent to the removal of the MYC "brake", which accelerates the growth of cancer.
In May, Nature Communications reported that blocking MYC may be "unexpectedly effective" in treating MRT and other cancers caused by SNF5 inactivation.
"One difficulty in treating cancers like MRT is that it is driven by a specific protein loss in tumor cells," said Dr. William Tansey, professor of cell and developmental biology.
"If MYC is activated by SNF5 damage, it means it will be an ideal target for these cancers," he said. Tansey, the co-leader of this article, is an internationally renowned expert in the MYC field.
The MYC family has three protein members, and in the United States, up to 100,000 cancer patients die from MYC each year.
MYC proteins act as transcriptional regulators, controlling the expression of thousands of genes involved in cell growth, proliferation, metabolism, and genomic instability. Tansey Labs is committed to identifying the underlying mechanisms of MYC work and developing new strategies for MYC in the clinic.
SNF5 inhibits MYC recognition of target genes in MRT cells
Tansey first suspected whether SNF5 regulates the interaction of MYC with chromatin in the context of MRT, as loss of SNF5 causes the MYC target gene to be repeatedly activated. The researchers used shRNA-mediated MYC knockout (the lentiviral shRNA structure designed by Saiye) MRT cells to confirm the importance of MYC to MRT cells, thereby enhancing the association between SNF5 and MYC. Next, a system was established to compare the effects of reintroducing SNF5 and inhibiting MYC in MRT cells in the same environment. ChIP was combined with next-generation sequencing (ChIPSeq) to track the distribution of MYC in the G401 cell genome. In contrast to the EGFP-expressing control, the researchers found 900 MYC binding peaks, which in turn compared the effects of OmoMYC or SNF5 expression. The results showed that OmoMYC reduced detectable genome-wide MYC binding and, more importantly, SNF5 also reduced MYC binding, and the effect of SNF5 on MYC binding was genome-wide, with an average reduction in binding strength of approximately three-fold, but MYC Steady-state levels are not affected by SNF5 expression, excluding the possibility of a decrease in binding reduced by MYC protein.
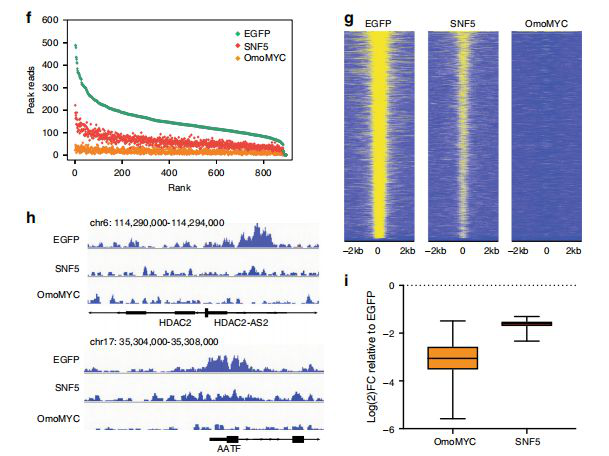
Using ATAC-seq, RNA-seq, and PRO-Seq, Tansey and colleagues demonstrated that the binding of SNF5 to MYC is independent of the role of SNF5 in chromatin remodeling, but because of the control of RNA polymerase in the MYC regulatory gene. The suspension is released. SNF5 selectively inhibits the binding of MYC to DNA and is required for its tumorigenesis.
These findings provide a new model for MRT tumorigenesis, in which deletion of SNF5 deactivates MYC. Therefore, reintroduction of SNF5 into MRT cells can displace MYC from chromatin, thereby inhibiting protooncogene expression.
Original Search: Inhibition of MYC by the SMARCB1 tumor suppressor
Fufeng Sinuote Biotechnology Co.,Ltd. , https://www.sinuotebio.com